Crouzon sendromu, Ayrıca Crouzon hastalığı adı verilen, kraniyal sütürlerin erken kemikleştiği, kafatasının büyümesinin bozulduğu ve kafa ve yüzde tipik malformasyonlar ve yapışıklıklar meydana gelebildiği bilinen, genetik olarak neden olunan birkaç kraniyosinostozdan biridir. Crouzon sendromundan etkilenen kişilerin zihinsel gelişimi genellikle normaldir.
Crouzon Sendromu nedir?

© royaltystockphoto - stock.adobe.com
Crouzon sendromu da Kraniyofasiyal dizostoz Crouzon adı verilen, bilinen birkaç kraniyosinostozdan biridir. Hastalık, bazıları prenatal olarak başlayan kraniyal sütürlerin erken kemikleşmesi ile karakterizedir. Ossifikasyon, büyüme aşamasında beynin normalde "çocukla birlikte büyüyen" takke altına kolayca yayılamayacağı anlamına gelir. Bunun yerine, takke esas olarak henüz kemikleşmemiş kraniyal sütürler üzerinde büyür, böylece tedavi edilmezse tipik malformasyonlar meydana gelir.
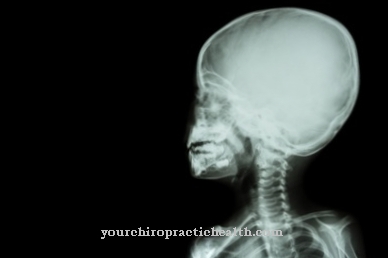
Crouzon sendromunda, başlangıçta koroner, alfa ve sagital sütürler kemikleşir. Tedavi olmaksızın veya düzeltici cerrahi müdahaleler olmadan, tipik bir kule kafatası ve aşırı göz rahatlaması (hipertelorizm) ve çıkıntılı gözler (ekzoftalmi) gibi yüz anomalileri ortaya çıkar. Crouzon sendromunda dişlerin yanlış hizalanmasına ek olarak sağırlık beklenmelidir çünkü dış işitsel kanalda tıkanıklık vardır, işitme kanalı atrezisi vardır ve / veya kemikçikler tam olarak gelişmediğinden işitme bozukluğu oluşur.
nedenleri
Crouzon sendromuna yalnızca kromozom 10 üzerindeki 10q26 gen lokusundaki bir mutasyon neden olur. İnsan hücre DNA'sının% 4 ila% 4,5'ini içeren 10. kromozomda yaklaşık 1.200 gen vardır. 10q26 geni, “fibroblast büyüme faktörü reseptörü 2” nin (FGFR2) kodlanmasından sorumludur. Bu spesifik gen mutasyonunun etkileri, gözlemlenen bir aralık içinde farklı şekilde telaffuz edilir.
Gen mutasyonu, otozomal dominant bir özellik olarak kalıtılır. Bu, Crouzon sendromunun cinsiyete özgü olmadığı, dolayısıyla kadınları ve erkekleri eşit şekilde etkileyebileceği anlamına gelir ve bu, 10q26 lokusundaki genetik kusurdan yalnızca bir ebeveyn etkilense bile hastalığın kesinlikle ortaya çıkacağı anlamına gelir. Bu genetik bozukluğun en dikkat çekici etkisi kafatası dikişlerinin erken kemikleşmesidir. Kraniyal sütürler, frontal kemiğin (Os frontale), parietal kemiğin (Os parietale) ve oksipital kemiğin (Os oksipitale) kemik plakalarının büyüme plakalarını temsil eder.
Sütürler büyüme aşamasında kemikleşirse, kafatası düzgün bir şekilde genişleyemez ve beyin büyüme basıncının artmasına neden olur, bu da kafatasının tipik deformasyonlarına yol açar.
Belirtiler, rahatsızlıklar ve işaretler
Kule kafatası, çıkıntılı gözler ve geniş göz rahatlığı gibi yukarıda açıklanan Crouzon sendromunun özellikle dikkat çekici semptomlarına ek olarak, Crouzon sendromunun varlığını gösteren başka işaretler de vardır. Bunlar, çıkıntılı kemikleşmiş kafatası sütürleri, gözlerin şaşı pozisyonu ve şaşılıktır. Şaşılık, göz kaslarının koordinasyon eksikliğidir.
Gözler ortak bir nesneye paralel veya hizalı olamaz. Üst çene hipoplazisi ve çıkıntılı bir alt dudak da Crouzon sendromunun yan etkileridir. Maksiller retrognati olarak da bilinen maksiller hipoplazinin semptomatik özelliği, çenenin maksilladan uzaklaşmasıdır.
Genel olarak sonuç, içbükey bir yüz ifadesinin görüntüsüdür. Kural olarak, Crouzon sendromunun semptomatik belirtileri kafatası ile sınırlı değildir, ancak diğer "ilişkili" problemler ortaya çıkar. Humero-radyal sinostozdan, omuz ekleminde kısmi bir ossifikasyondan ve dirsek ekleminde bir subluksasyondan bahsedilmelidir.
Teşhis ve kurs
Crouzon sendromunun olası bir varlığı şüphesi, aile geçmişine bağlı olarak doğum öncesi olarak ortaya çıkabilir. Dışarıdan görülebilen semptomlar, tanısal görüntülemeyi neredeyse gereksiz kılar. Crouzon sendromunun mevcut olup olmadığına dair herhangi bir şüpheniz varsa, genetik bir analiz bilgi sağlayabilir. Hastalığın seyri, özellikle ilişkili klinik tablolarda kişiden kişiye değişir.
Hastalık tedavi edilmezse, ana semptomlar kafatasının ve beynin ana büyüme aşamasında ortaya çıkma eğilimindedir. Büyüme evresi sona erdikten sonra baş ve yüzde açıkça görülebilen deformasyonlar, cerrahi müdahaleler yapılmadıkça ömür boyu kalır.
Komplikasyonlar
Kural olarak, Crouzon sendromunda özellikle kafatasının oluşumu ciddi şekilde etkilenerek kafatası sütürlerinin kemikleşmesi ile sonuçlanır. Bu, kafada hastanın görünümünü büyük ölçüde etkileyen muazzam şekil bozukluklarına neden olabilir. Çoğu insan, özgüveninin azalmasından muzdariptir ve kendini çekici hissetmez.
Crouzon sendromu, özellikle çocuklarda ve gençlerde olduğu gibi sosyal zorluklara da yol açabilir. Değişen görünüm zorbalığa yol açabilir. Crouzon sendromunda, çoğu durumda zihinsel gelişim bozulmaz. Gözler ayrıca Crouzon sendromundan etkilenir, böylece bir şaşılık meydana gelebilir. Bu, koordinasyonda zorluklara yol açar.
Crouzon sendromu çocuklukta cerrahi olarak tedavi edilmediğinde komplikasyonlar ortaya çıkar. Tedavinin kendisi ancak cerrahi bir önlem olarak mümkündür ve öncelikle malformasyonları düzeltmeyi amaçlamaktadır. Her şeyden önce büyüyen beyin için alan yaratılır. Bununla birlikte, sendromun tam bir tedavisi mümkün değildir. Operasyon sırasında belirli bir komplikasyon olmadığı sürece yaşam süresinde azalma olmaz.
Ne zaman doktora gitmelisiniz?
Çoğu durumda, Crouzon sendromu doğumdan hemen sonra veya hatta doğumdan önce tanınır, böylece çoğu durumda ek bir teşhis gerekli değildir. Ancak bireysel şikayet ve şekil bozukluklarının tedavi edilebilmesi için doktora başvurulmalıdır.
Özellikle çocuğun şaşkınlığı varsa bu şikayeti düzeltmek için doktora başvurulmalıdır. Yüzdeki kaslar da Crouzon sendromundan etkilenebilir, bu nedenle hasta bağımsız bir yüz ifadesi oluşturamıyorsa doktora başvurmak gerekir.
Eklemlerin kemikleşmesi de sendromu gösterebilir ve incelenmesi gerekir. Kural olarak, semptomlar bir çocuk doktoru veya bir pratisyen hekim tarafından incelenip teşhis edilebilir. Daha ileri tedavi semptomların ciddiyetine bağlıdır, bu nedenle cerrahi müdahaleler gerekli olabilir.
Crouzon sendromuna bağlı olarak çocuk veya akrabaları ve ebeveynleri psikolojik şikayetler yaşarsa, daha fazla şikayet ve komplikasyondan kaçınmak için bir psikoloğa da danışılmalıdır.
Bölgenizdeki doktorlar ve terapistler
Tedavi ve Terapi
Crouzon sendromunun tedavisi esasen - eğer varsa - düzeltici cerrahiden oluşur. Uzman klinikler tarafından sunulan bilinen üç farklı cerrahi teknik vardır. Ön yörünge ilerlemesi, kafatasında alın da dahil olmak üzere kafatasının önünün kesilmesi ve beynin gerekli büyüme için yer açması için yeniden sabitlenmesinden oluşur.
Kafatasının yeniden tutturulması temel olarak titanyum plakalar, emilebilir plak sistemi veya emilebilir sütür materyali ile yapılabilir. Hangi yöntemin kullanılacağı, operasyon sırasında karşılaşılan koşullara bağlıdır. Kemikli yüz kafatasındaki operasyonlar genellikle daha karmaşıktır ve Le Fort III osteotomisi olarak bilinir. Bazı durumlarda bu, çok geniş bir göz pozisyonunu da düzeltebilir.
Üçüncü prosedür, distraksiyon osteogenezi, kafatası plakalarının kademeli olarak yer değiştirmesine izin verir. Kafatasının belirli bölgelerine yönelik distraksiyon cihazları cerrahi olarak kullanılır ve operasyondan sadece birkaç gün sonra yerleşik fiksasyon sistemi kullanılarak kemik plakları her gün bir milimetreye kadar birbirinden çıkarılabilir. Kemik, boşluğu daha sonra kemikleşen ve bir tür yapay kafatası büyümesi oluşturan nasır dokusuyla doldurur.
Görünüm ve tahmin
Crouzon sendromu her zaman tedavi edilmelidir. Sendrom tedavi edilmezse genellikle ölüme yol açar. Tedavi sadece sendromun semptomlarına dayanabilir ve nedensel olamaz.
Oluşumlar cerrahi müdahaleler ile düzeltilir. Erken teşhis ve tedavi, hastalığın ilerleyişinde çok olumlu bir etkiye sahiptir. Erken bir operasyon, beyne sağlıklı büyüme için yeterli alan sağlar, böylece hastanın yaşamında daha fazla kısıtlama veya rahatsızlık olmaz.
Kural olarak, ilgili kişi işlemden sonra başka bir şikayette bulunmaz ve herhangi bir komplikasyon söz konusu değildir. Erken tedavi edilirse hastanın zihinsel gelişimi de hastalıktan rahatsız olmaz. Başarılı bir tedaviden sonra bile, başka semptomlardan kaçınmak için düzenli muayeneler tavsiye edilir.
Kural olarak, Crouzon sendromu, erken keşfedilir ve tamamen tedavi edilirse, etkilenen kişinin yaşam beklentisi üzerinde olumsuz bir etkiye sahip değildir. Crouzon sendromu tedavi edilmezse, yüzde daha fazla malformasyona ve dolayısıyla etkilenen kişinin yaşamında kısıtlamalara yol açacaktır. Özellikle omuz eklemleri ve gözler etkilenir.
Önlemek
Crouzon sendromunun oluşumu genetik olduğundan, doğrudan önleyici tedbirler bilinmemektedir. Hastalığı tek başına önleyecek tedbirler yoktur. Bununla birlikte, nedensel genetik kusur şüphesi varsa, önleyici tedbirler, özellikle büyüme aşamasında, hastalığın etkilerini en aza indirmek için önemlidir.
Önleyici tedbirler, kafatasına cerrahi müdahale ile beyne kafatasının altında normal büyüme imkânı sunmak için kafatasının düzenli görsel muayenesi ve kafa içi basıncının incelenmesinden oluşur.
tamamlayıcı tedavi
Crouzon sendromu ile çoğu durumda, etkilenenler için özel bir takip önlemi yoktur. Bu hastalıkta, hasta, daha fazla komplikasyondan veya semptomların daha da kötüleşmesinden kaçınmanın tek yolu olduğundan, öncelikle hızlı bir tanıya ve sonraki tedaviye bağımlıdır.
Bu nedenle, hastalığın ilk belirtilerinde bir doktora danışılmalıdır. Tedavi ne kadar erken başlarsa, bu hastalığın ilerideki seyri genellikle o kadar iyi olur. Bu genetik bir hastalık olduğundan, çocuk sahibi olmak istiyorsanız her zaman önce genetik danışma yapılmalıdır. Bu, Crouzon sendromunun torunlarda tekrar ortaya çıkmasını önleyebilir.
Tedavi cerrahi bir prosedürle gerçekleştirilir. Etkilenen kişi bu işlemden sonra mutlaka dinlenmeli ve vücuduna iyi bakmalıdır. Vücudu gereksiz yere zorlamamak için efordan, stresli ve fiziksel aktivitelerden kaçınmalısınız. Dahası, kişinin kendi ailesinden veya arkadaşlarından ve tanıdıklarından aldığı destek genellikle çok önemlidir. Çoğu durumda, bu hastalık etkilenen kişinin yaşam beklentisini azaltmaz.
Bunu kendin yapabilirsin
Crouzon sendromlu bir çocuk doğduğunda, ilk meydan okuyan ebeveynlerdir. Hastanın kapsamlı bir teşhis geçirmesi önemlidir. Hem cerrahi müdahaleleri hem de tıbbi yardım seçimini içeren tedavi planları oluşturulabilir. Özellikle kafatası operasyonları olabildiğince erken yapılmalıdır.
Bununla birlikte, Crouzon sendromlu hastalar yalnızca kapsamlı tıbbi tedaviye ihtiyaç duymazlar. Genellikle erken yaşlardan itibaren bakılır, marjinalleştirilir veya zorbalığa uğrarlar. Ebeveynlerin ve kardeşlerin de dahil edilmesi gereken empatik psikoterapi burada yardımcı olabilir.
Ebeveynlerin ve hastaların etkilenen diğer kişilerle temasa geçmesi genellikle yararlıdır. Bunun için birkaç olasılık var.Örneğin, "Ebeveyn Girişimi Apert Sendromu ve İlgili Malformasyonlar e.V." nin (www.apert-syndrom.de) web sitesi de Crouzon sendromu hakkında bilgi sağlar ve ayrıca aile birleşimi olarak yıllık bir eğitim etkinliği sunar.
Crouzon sendromlu yaklaşık seksen hasta şu anda "Diseasemaps" web sitesinde kayıtlıdır. Bu saygın siteye katılırsanız, ilgili ülkelerdeki bireysel hastalarla iletişime geçebilirsiniz (www.diseasemaps.org/de/crouzon-syndrome).
Sendrom otozomal dominant bir şekilde miras alındığı için, Crouzon sendromlu hastalar gebe kalmaya çalışırken genetik tavsiye almalıdır.

.jpg)

.jpg)



.jpg)


.jpg)