Joubert sendromu beyin sapında konjenital bir malformasyon ve ayrıca bir agenez (inhibisyon malformasyonu, bağlanma eksikliği, örneğin serebral barlar, apendiks) ile karakterize edilir. Serebellar solucanın hipoplazisi (az gelişmişliği) de mevcut olabilir. Bu otozomal resesif genetik kusurdan muzdarip hastalar, diğer şeylerin yanı sıra anormal solunum davranışı ve ataksi gösterir.
Joubert Sendromu nedir?

© Sashkin - stock.adobe.com
İle insanlar Joubert sendromu merkezi sinir sistemi gelişimsel bozuklukları ve bunun sonucunda ortaya çıkan fonksiyonel bozukluklardan muzdariptir. Tıbbi araştırmalar, bu genetik bozukluğun kendi başına bir hastalık olarak sınıflandırılıp sınıflandırılmayacağı konusunda tartışmalıdır.
Etkilenen hastaların çeşitli farklı semptomları vardır. Bu nedenle kesin teşhis zordur. JB, geniş gen lokus heterojenliği ile karakterizedir. Şimdiye kadar çoklu gen mutasyonları tespit edildi. Bir mutasyon analizi çok kapsamlıdır.
nedenleri
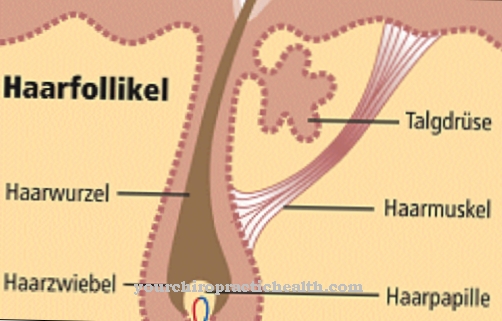
Joubert sendromu, birincil siliyofati grubuna aittir. Birincil kirpikler veya bazal bedenin bu genetik bozukluğu ile farklı gelişimsel bozukluk türleri ortaya çıkabilir. Kirpikler, özel hücre işlemleri olarak çeşitli görevleri yerine getirir. Kemo, mekano ve ozmoz sensörleri olarak hareket ederler ve birçok sinyal yoluna dahil olurlar. Ayrıca normal organ gelişimini sağlarlar.
Temel gelişim süreçlerinin doku homeostazını sürdürürler. İlgili proteinlerin büyük bir kısmı etkileşim yoluyla karmaşık bir ağ oluşturur. Ana semptomlara ek olarak başka organlar da etkilenirse, JSRD (Joubert Sendromu İlişkili Bozukluk) mevcuttur. Bu ikincil hastalık böbrekleri, karaciğeri ve gözleri içeren başka organ belirtileriyle karakterizedir.
Genetik olarak heterojen bir sendromdur. Doktorlar, NPHP6 / CEP290 geninde (nefrosistin-6 için kodlama) veya NPHP8 / RPGRIP1L geninde (nefrokistin-8 için kodlama) malformasyonlar buldular. Diğer gen mutasyonları MKS3, ARL13B, AHI1, CC2DA2, TMEM216 ve INPP5E'dir. Yalnızca birkaç hastada NPHP4 ve NPHP1'de mutasyonlar vardır.
Belirtiler, rahatsızlıklar ve işaretler
Patognomonik özellik, "eksenel T1 ağırlıklı beyin manyetik rezonans görüntüleme" kullanılarak belirlenebilen "molar diş işaretidir" (MTS). Bu özellik, serebellar solucan veya serebellar solucanın agenezisi veya hipoplazisi ile karakterizedir. Ayrıca, posterior interpendiküler fossa (serebral bacaklar arasındaki çukur) güçlü bir şekilde içeri çekilir ve orta beyindeki bir malformasyon nedeniyle serebellar saplar belirgin bir üstün şekle sahiptir.
MTS'ye ek olarak, hastalar sıklıkla solunum bozuklukları, ataksi, kas hipotansiyonu ve psikomotor gerilikten muzdariptir. Etkilenenlerin yüzde 8 ila 19'u postaksiyal polidaktili (çoklu parmak) ve yüzde 6'sında oksipital (meningo) ensefalosel var, burada beynin arkası şişkin.
Bu deformite ilk olarak 1969'da kaydedildi. Prevalans, hastalığın ne kadar seyrek ortaya çıktığını gösteren bir oran olan yaklaşık 1: 100.000'dir. İlk tıbbi araştırmadan bu yana sadece yüz vaka belgelendi. Bu genetik kusur farklı şekillerde ve varyantlarda meydana geldiğinden, doktorlar genetikte çok sayıda değişiklik olduğunu varsayarlar.
Kesin bir anormallik henüz kesin olarak doğrulanmadı. Bununla birlikte, X kromozomunun bir mutasyonu kesin kabul edilir. Bu bozukluk otozomal resesif kalıtım temelinde geçmektedir. Eksik bir vermis serebelli (serebellum, serebellar kurt), retinada hasar ve gözle görülür bir iris söz konusudur.
Yenidoğan döneminde sıklıkla ortaya çıkan semptom ve şikayetler, nistagmus ve epizodik taşipne ve apne gibi düzensiz solunum paternidir. Küçük çocuklar hipotoni geliştirebilir. İlerleyen yaşla birlikte dengesizlik ve düzensiz bir yürüyüş gelişir (ataksi). Bu ana semptomlar aynı zamanda motor kilometre taşları olarak da bilinir.
Hastaların farklı seviyelerde bilişsel yetenekleri vardır ve ciddi şekilde bozulabilir, ancak aynı zamanda normal bir zeka seviyesi de gösterebilirler. Okülo-motor apraksi (hareket bozukluğu) da mümkündür.
Bu genetik kusurun özelliği, büyük bir kafa, yuvarlak ve yüksek kaşlar, belirgin (çıkıntılı) bir alın, deforme olmuş bir ağız, ritmik olarak hareket eden ve çıkıntı yapan bir dil ve derin kulaklar gibi kraniyofasiyal anormalliklerdir. Ara sıra görülen semptomlar nefrofthisis, retina distrofi ve polidaktilidir.
Hastalığın teşhisi ve seyri
Gebeliğin 18. haftasından sonra ataksi, hipotansiyon, okülomotor apraksi, açık vermis serebelli ve gelişimsel gecikmenin daha önce belirtilen karakteristik kilometre taşlarına dayanarak bir tanı konur. Ek olarak, MRI'da, MTS'de (Molar diş işareti) karakteristik bir nöroradyolojik bulgu yapılır.
Molar işaret olarak bilinen bu özellik, pastil ve orta beyin kusurlarının yanı sıra küçük beyin solucanının hipoplazisinden kaynaklanmaktadır. JSRD (Joubert sendromu ile ilgili bozukluk), Dandy-Walker malformasyonu (MTS'siz malformasyonlu serebellar solucan), okülomotor apraksi tip 1 ve 2, ponto-serebral hipoplazi ve atrofi, 3-c gibi JS ile yakından ilgili hastalıklar temelinde farklı teşhisler yapılır. Sendrom, orofacio-dijital sendromlar II ve III ile Meckel-Gruber sendromu.
Aşama I, JBTS5 (53 kodlama eksonu), JBTS3 (26 kodlama eksonu), JBTS6 (28 kodlama eksonu) ve JBTS9 (36 kodlama eksonu) genlerinin “yeni nesil dizileme tabanlı panel analizi” ni içerir. JBTS4 geni multipleks PCR ile homozigot delesyon için test edilir. Aşama II'de, diğer JB genleri, azalan mutasyon frekanslarına karşılık gelen fenotipik özelliklere bağlı olarak PCR (enzime bağlı olarak DNA zincirindeki gen dizilerini kopyalayan bir işlem) ve ardından Sanger dizilimi ile analiz edilir.
Kromozom dengesizliklerini ortadan kaldırmak için, diferansiyel teşhis SNP dizisi analizi yapılır. Bir akrabalık varsa veya aile içinde birkaç hasta biliniyorsa, doktorlar homozigotluk taramasını, geni çevreleyen mikro uydu işaretleyicisinde birleştirme analizi ve ardından Sanger dizilemesi kullanarak gen analizi yoluyla gerçekleştirir. Tanı materyali olarak çocuklardan iki ila on mililitre EDTA kanı alınır; yetişkinlerden bu miktar beş ila on mililitredir.
DNA veya doku malzemesi de uygundur. Aşama I: Genomik DNA materyali, MLPA kullanılarak NPHP1 geninin kantitatif bir analizi yoluyla duplikasyonların veya delesyonların varlığı açısından incelenir. Genomdaki çok küçük miktarlarda DNA, tek tek eksonların (gen segmentleri) silinmeleri ve kopyalanmaları için incelenir. Evre II: Şimdiye kadar tanımlanan genlerin kodlanmış eksonları, yeni nesil frekanslar kullanılarak değerlendirilir. Ek yerleri, prob hibridizasyonu ile zenginleştirilir.
Komplikasyonlar
Joubert sendromu, çoğu hastanın çeşitli rahatsızlıklardan muzdarip olmasına neden olur. Bu genellikle boy kısalığına, solunum bozukluklarına ve ayrıca gecikmeye yol açar. Çocuğun zihinsel gelişimi de kısıtlanabilir. Nefes darlığı da kesinlikle tedavi edilmesi gereken nefes darlığına neden olabilir.
Kişinin ebeveynlerinin şiddetli depresyon veya diğer psikolojik bozukluklardan muzdarip olması nadir değildir. Hastalar ayrıca denge bozuklukları gösterirler ve sıklıkla kısıtlı hareketlilikten muzdariptirler. İşitme kaybına veya görme sorunlarına yol açan gözlerde ve kulaklarda rahatsızlık nadir değildir. Hastanın yaşam kalitesi Joubert sendromu ile önemli ölçüde azalır.
Çeşitli terapilerin yardımıyla Joubert sendromu kısıtlanabilir ve tedavi edilebilir. Maalesef nedensel bir tedavi yapılamaz. Acil durumlarda, nefes darlığı varsa acil ventilasyon da yapılabilir. Tedavinin kendisinde belirli bir komplikasyon yoktur. Genel olarak, hastanın yaşam beklentisinin Joubert sendromu tarafından azaltılıp azalmayacağı tahmin edilemez.
Ne zaman doktora gitmelisiniz?
Hamile bir anne, hamilelik sırasında mevcut tüm kontrollere katılmalıdır. Muayenelerde hem hamile kadının hem de doğmamış çocuğun sağlık durumu incelenir. Joubert sendromu, hamileliğin 18. haftası kadar erken teşhis edilebildiğinden, sağlık sigortası şirketleri tarafından önerilen önleyici tıbbi kontrollerden yararlanmanız önerilir. Ayrıca ebeveyn atalarının geçmişinde genetik bir kusur varsa, genellikle genetik danışma ve muayene önerilir.
Rahimde herhangi bir düzensizlik bulunmaması durumunda, doğumdan hemen sonra kadın doğum uzmanları ve pediatristler tarafından otomatik kontroller yapılır. Bu muayeneler sırasında solunum bozuklukları tespit edilebilir. Çocuğun ebeveynleri daha önce fark edilmeyen herhangi bir olağandışı tutarsızlık fark ederse, gözlemler bir doktorla tartışılmalıdır. Herhangi bir fiziksel özellik, boy kısalığı veya deformiteler varsa bir doktora danışılmalıdır.
Aynı yaştaki çocuklarla doğrudan karşılaştırıldığında, dil sorunları veya zihinsel yetersizlik fark edilirse, bir doktora danışılmalıdır. Nedeni açıklığa kavuşturmak için soruşturmalar gereklidir. Ne kadar erken teşhis konulursa, çocuğu desteklemek için o kadar erken hedeflenen tedaviler başlatılabilir. Bu nedenle, bir anormalliğin ilk belirtisinde bir doktora danışılmalıdır.
Tedavi ve Terapi
Ebeveynlerin genetik danışmanlık alma hakları vardır. Tedavi seçenekleri, bu hastalığın nedenleri çok çeşitli olduğu kadar çeşitlidir. Motor gelişim bozuklukları ve hipotansiyon durumunda, hastalığın seyrini olumlu yönde etkileyebilecek eğitim destek programları, dil, mesleki ve mesleki terapi devreye girer.
Anormal solunum paternlerinden etkilenenlere oksijen ikamesi veya ventilasyon da verilebilir. Hafif semptomları olan hastaların prognozu pozitiftir. Ciddi şekilde etkilenen hastalar, uzman bir referans merkezi tarafından bakılmalıdır.
Görünüm ve tahmin
Joubert sendromunun prognozu kötü. Bu sendrom genetik bir bozukluktur. Mevcut tıbbi, bilimsel ve yasal gerekliliklerle bu tedavi edilemez. Araştırmacıların ve doktorların, bir kişinin genetik koşullarını müdahalelerle değiştirmesine yasal olarak izin verilmez. Bu nedenle tedavi, mevcut yaşam kalitesini iyileştirmeye yönelik tedavilerin kullanımına yöneliktir. Tıbbi bakım kullanılmadan, hastanın azalan refahı daha da azalır.
Sendrom ne kadar erken teşhis edilir ve tedavi edilirse sonuçlar o kadar iyi olur. Acil durumlarda ilgili kişinin acil ventilasyonu endikedir, aksi takdirde hasta erken ölebilir. Bireysel bir tedavi planında çok sayıda tedavi bir araya getirilip uygulanmasına rağmen, mevcut hastalık ikincil bozukluklara yol açabilir. Bunlar genel prognozu kötüleştirir.
Mevcut işlevsel bozukluklar veya hareket üzerindeki diğer kısıtlamalar akıl hastalıklarına yol açabilir. Geçici veya kalıcı depresyon, duygudurum dalgalanmaları veya kişilikteki değişiklikler birçok hastada belgelenmiştir. Bu, ilgili kişi ve çevre için ek bir yükü temsil eder Joubert sendromlu bir hastanın günlük yaşamı, genellikle ancak akrabalarının yeterli yardımı ve desteği ile yönetilebilir. Denge bozuklukları ve ataksi yaşla birlikte daha şiddetli hale gelir.
önleme
Kesin bir genetik nedensellik henüz kesin olarak belirlenmediğinden, klinik anlamda önleyici tedbirler yoktur. İnsan organizmasındaki malformasyonları önlemenin tek yolu sağlıklı bir yaşam tarzı sürdürmektir.
tamamlayıcı tedavi
Çoğu durumda, Joubert sendromlu hastanın doğrudan veya özel takip seçenekleri yoktur, bu nedenle etkilenen kişi öncelikle hastalığın hızlı ve her şeyden önce erken teşhisine bağımlıdır. Hastalık ne kadar erken fark edilirse, daha sonraki seyir genellikle o kadar iyi olur. Bu nedenle, ilk belirti ve bulgularda bir doktora başvurmanız tavsiye edilir.
Bu hastalıkta, etkilenen kişi genellikle semptomları hafifletebilecek yoğun bakım ve tedaviye bağımlıdır. Etkilenen kişinin mümkün olduğunca normal bir yaşam sürmesini sağlamak için ebeveynlerin ve yakın akrabaların yardım ve desteği de çok talep görmektedir. Genellikle fizyoterapi veya fizyoterapi egzersizleri kendi evinizde de yapılabilir, bu da semptomları hafifletebilir.
Belirtiler her zaman tamamen ortadan kaldırılamaz. Diğer Joubert sendromu hastaları ile iletişim kurmak da çok yararlı olabilir, çünkü bilgi alışverişinde bulunmak alışılmadık bir durum değildir. Kural olarak, etkilenen kişinin yaşam beklentisi bu hastalık tarafından azaltılmaz.
Bunu kendin yapabilirsin
Joubert sendromu tedavi edilemez ve günlük yardım da zordur. Doğuştan hastalığın semptomları çoğu durumda kaçınılmazdır. Yine de bazılarının hafiflemesi olasıdır.
Etkilenen kişilerde solunum özellikle rahatsız olduğu için bu bir başlangıç noktasıdır. Optimize edilmiş bir oda iklimi yardımcı olabilir. Kuru ısıtma havası, solunum sorunlarını şiddetlendirebilir. Çok soğuk hava da aynı etkiye sahiptir. İdeal olarak, oda sıcaklığı yaklaşık 20 ° C ve nem oranı yaklaşık yüzde 50'dir. Özellikle iç mekan bitkileri, optimum bir iç mekan iklimine katkıda bulunabilir. Alternatif olarak, nemi istenen seviyede tutmak için odaya nemli havlular da yerleştirilebilir. İç ortam iklimi bir higrometre kullanılarak izlenebilir. Nefes almayı da hedefleyen bir başka başlangıç noktası nefes egzersizleridir. Düzenli kullanım, aksi takdirde otomatik olan sürecin algısını geliştirir. Bu şekilde çok hızlı nefes almayı ve nefes almanın duraklamalarını engelleyebilirsiniz.
Ayrıca, etkilenenlerin bir odada yalnız uyumaması da mantıklıdır. Akrabalar uyku sırasında nefes almada duraklamalar fark edebilir ve hastayı uyandırabilir veya nefes almaya teşvik edebilir. Ama bu sadece bir önlem.
.jpg)










.jpg)