Maroteaux-Lamy Sendromu çeşitli lizozomal depo hastalıklarını içeren mukopolisakkaridozlara aittir. Sendrom, yetersiz enzim aktivitesiyle sonuçlanan ve dermatosülfat birikimlerine yol açan genetik bir mutasyona bağlıdır. Terapi, esas olarak enzim replasman tedavilerinden oluşur.
Maroteaux-Lamy Sendromu nedir?

© imaginuma - stock.adobe.com
Mukopolisakkaridozlar, lizozomal depo hastalıkları içeren bağımsız bir hastalık grubudur. Lizozomal depo hastalıkları köşede var. Hepsi, lizozomun arızalanmasının neden olduğu genetik olarak belirlenmiş metabolik hastalıklardır. Bu hastalıklardan biri sözde Maroteaux-Lamy Sendromu. Doğuştan gelen metabolik bozukluk, dermatinsülfatların depolanmasına yol açar.
Terimler eşanlamlı olarak kullanılmaktadır Mukopolisakkaridoz tip VI, Aril sülfataz B eksikliği, ARSB eksikliği ve ASB eksikliği gibi N-asetilgalaktozamin-4-sülfataz eksikliği. Hastalık ilk olarak 1963'te tanımlandı. Parisli insan genetikçiler ve pediatristler P. Maroteaux ve M. Lamy, hastalığı ilk tanımlayanlar olarak kabul ediliyor.
Hastalığın prevalansı, 100.000 kişide bir ila dokuz hasta arasındadır. Şimdiye kadar belgelenen vakalarda aile birikimi görüldü. Sendrom, otozomal resesif bir şekilde kalıtılır. Hastalığın nedeni genetik bir mutasyon olarak kabul edilir.
nedenleri
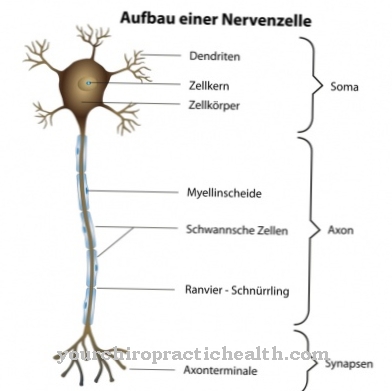
Maroteaux-Lamy sendromu, çoğunlukla kalıtsal bir mutasyona bağlıdır. Nedensel mutasyonlar artık ARSB geninde lokalize edilmiştir. 5q13 ila 5q14.1 gen lokusundaki mutasyonların semptom kompleksine neden olabileceği söylenir. Orada bulunan genler, belirli bir proteini DNA içinde kodlar.
Genlerin bir mutasyonunun, ASB veya N-asetilgalaktozamin-4-sülfataz olarak da bilinen mutasyona uğramış aril sülfataz B'nin anormal aktivitesine yol açtığı söylenir. Mutasyonun bir parçası olarak maddenin aktivitesi azalır. Bu azalma nedeniyle, kondroitin sülfat ve dermatan sülfat gibi maddelerin parçalanmasında aksamalar yaşanmaktadır. Maddeler artık nedensel mutasyon nedeniyle yeterince parçalanmadığından vücut, maddelerin kalıntılarını depolar.
Maroteaux-Lamy sendromunun tipik semptomları bu depolamanın bir sonucudur. Mutasyonun gelişiminde genetik faktörlerin olduğu kadar dış etkilerin de rol oynayıp oynamadığı henüz bilinmemektedir. Bu, en azından yeni mutasyonlar için varsayılabilir.
Belirtiler, rahatsızlıklar ve işaretler
Maroteaux-Lamy sendromlu hastalar, klinik olarak karakteristik bir kriter kompleksinden muzdariptir. En önemli semptomlardan biri, orantısız bir şekilde hareket eden kısa boydur ve kısa bir gövde ile karakterize edilir. Hastanın orantısızlığı, Hurler hastalığının semptomlarını anımsatan kaba bir yüzle ilişkilidir.
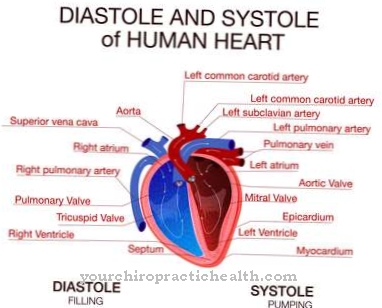
Çoğu durumda, hastanın korneası bulanıktır. Ek olarak, hepatosplenomegaller, fıtıklar veya eklemlerin kontraktürleri olabilir. Tortular nedeniyle hastaların kalp kapakçıkları giderek kalınlaşır. Ek olarak, disostoz multipleksli hastalardakine benzer iskelet displazisi olabilir.
Hem klinik tablo hem de sendromun seyri değişken ve bireysel olarak kabul edilir. Yavaş süreçlere ek olarak, hızlı süreçler de belgelendi. İlk belirtiler doğumdan hemen sonra kendini gösterirse, bu fenomen oldukça hızlı bir seyir için konuşur.
Bu, özellikle idrardaki glikozaminoglikandaki artış, şiddetli dizostoz multipleks için ve kısa boy göstermesi için geçerlidir. Yavaş seyreden etkilenen kişiler genellikle ilk semptomları çok daha sonra gösterir. GAG artışı çok daha düşüktür ve disostoz multipleks çok daha hafiftir.
Hastalığın teşhisi ve seyri
Maroteaux-Lamy sendromunun doğumdan hemen sonra kendini göstermesi gerekmez ve çoğu durumda ancak daha sonra, ilk semptomlar ortaya çıktığında teşhis edilir. Teşhis, hastalığın klinik olarak tipik kriterlerine dayanmaktadır ve bu nedenle esas olarak kültürlenmiş fibroblastlar ve lökositler üzerinde izlenebilen önemli ölçüde azaltılmış ASB aktivitesine dayanmaktadır.
Öte yandan, diğer sülfatazlarla ilişkili olarak normal aktivite mevcuttur. Teşhisin bir parçası olarak, doktor ayrıca idrarla atılan dermatan sülfat miktarının arttığına dair kanıt sağlar. Ayırıcı tanıda Maroteaux-Lamy sendromu, çoklu sülfataz eksikliği ve diğer mukopolisakkaridoz formlarından ayırt edilmelidir. Siyalidoz ve mukolipidoz da ayırıcı tanı ile ilgili hastalıklardır. Hastanın prognozu vakadan duruma değişir ve öncelikle başlangıç yaşına ve ilk semptomların ciddiyetine bağlıdır.
Komplikasyonlar
Birincisi ve en önemlisi, Maroteaux-Lamy sendromu hastada kısa bir boy olmasına yol açar. Özellikle çocuklar, genç yaşta zorbalık ve alaydan muzdarip olabilir ve bunun sonucunda psikolojik sorunlar veya depresyon geliştirebilirler. Kural olarak, hastanın daha fazla büyümesi orantılı olarak gerçekleşmez ve yüzde de meydana gelen çeşitli şikayetler ve malformasyonlar meydana gelir.
Ayrıca, kalp kapakçıkları Maroteaux-Lamy sendromu tarafından hasar görür ve yanlış yerleştirilir, böylece kalpte şikayetler veya sınırlamalar olur. Bazı durumlarda, hastanın yaşam beklentisini de azaltabilir ve bu da kalp ölümüne neden olabilir. Bu sendromun nedensel tedavisi mümkün değildir. Bu nedenle tedavinin temel amacı, etkilenen kişinin sıradan bir yaşam sürdürebilmesi için semptomları sınırlandırmak ve bunlarla mücadele etmektir.
Psikolojik tedavi de gerekli olabilir. Tüm şikayetler tamamen sınırlı olmasa da belirli bir komplikasyon yoktur. Kural olarak, kalp semptomları yoksa Maroteaux-Lamy sendromu tarafından yaşam beklentisi de azalmaz.
Ne zaman doktora gitmelisiniz?
Büyüyen bir çocuk, fiziksel gelişim sürecinde rahatsızlık veya değişiklik belirtileri gösteriyorsa, bir doktora danışılmalıdır. Çocuk açıkça kısaysa veya iskelet sisteminde bir malformasyon varsa, tıbbi yardıma ihtiyacı vardır. Aynı yaştaki çocuklarla doğrudan karşılaştırıldığında vücut şeklinde anormallikler veya tuhaflıklar varsa, belirtileri netleştirmek için bir doktora danışılmalıdır. Hareket dizisi bozuklukları veya genel motor beceriler bir doktora sunulmalıdır. Kornea bulanıksa veya görme azalmışsa bir doktora da ihtiyaç duyulacaktır. Düzensiz kalp ritimleri, çeşitli tıbbi testlerle incelenmesi gereken bir sağlık sorununa işaret eder.
Çoğu durumda, iskelet sistemindeki değişiklikler nedeniyle hastalığın ilk anormallikleri doğumdan hemen sonra fark edilebilir. Bebekler doğduktan sonra hazır bulunan hekim tarafından kapsamlı bir şekilde muayene edildikleri için ebeveynlerin harekete geçmesine gerek yoktur. Yaşamın ilk birkaç haftası veya ayında kusma veya ağrı gibi belirtiler ortaya çıkarsa bir doktora danışılmalıdır. Çocuk sürekli ağlar ve ağlarsa, bu açıklığa kavuşturulması gereken bir düzensizliğin işaretidir. Nefes darlığı veya akut bir sağlık durumu durumunda, bir acil servis uyarılmalıdır. Gelene kadar çocuğun hayatta kalmasını sağlamak için ilk yardım sağlanmalıdır.
Tedavi ve Terapi
Maroteaux-Lamy sendromlu hastalar için daha dar anlamda nedensel bir terapi mevcut değildir, çünkü etkilenenlerin değişen enzim aktivitesi, yalnızca gen terapisi ile düzeltilebilecek bir genetik mutasyondan kaynaklanmaktadır. Bununla birlikte, en geniş anlamıyla enzim replasman terapisi ile, hastalığın semptomlarını kaynağında ele alan bir terapi türü mevcuttur.
Enzim replasman tedavisinde hastalar, naglazimler anlamında galsülfaz alırlar. Bu enzim ikamesi, ilgili maddelerin daha iyi parçalanmasıyla sonuçlanır ve böylece hastalığın seyrini geciktirir. Ancak şimdiye kadar meydana gelen semptomlar tamamen geri döndürülemez. Kalp kapakçığı kalınlaşması semptomatik olarak tedavi edilebilir ve belirli durumlarda kalp kapakçıklarının cerrahi olarak değiştirilmesini gerektirebilir.
Akut fıtıklar taksilerle tedavi edilir. Amaç, doktorun operatif bir çözüm bulması için zaman tanıyan bir azaltmadır. Körlüğe neden olan şiddetli kornea opasiteleri muhtemelen bir kornea nakli ile tersine çevrilebilir. Boy kısalığı gibi semptomlar nihayetinde geri döndürülemez, ancak genellikle bir enzim replasman ilacı ile erken tedavi ile sadece hafif bir formda ortaya çıkar.
İlaçlarınızı burada bulabilirsiniz
➔ Ağrı kesici ilaçlarGörünüm ve tahmin
Nadiren ortaya çıkan bu hastalık, çok çeşitli şekil ve şekillerde ortaya çıkar.Bireysel seyir ve farklı derecelerde Maroteaux-Lamy sendromu veya mukopolisakkaridoz tip 6 sayesinde, belirli bir hasta için güvenilir bir prognoz vermek genellikle zordur.
Genel olarak, Maroteaux-Lamy sendromunun ilk semptomları doğumdan kısa bir süre sonra ortaya çıkarsa prognoz daha kötüdür. Bu genellikle hastalığın daha hızlı seyri için konuşur.
Sonuç olarak, Maroteaux-Lamy sendromunun ilk semptomlarının ortaya çıktığı noktada olduğu gibi, etkilenenlerin yaşının da beklenen prognoz hakkında bir sonuca izin verdiği söylenebilir. Ayrıca hastanın görünümü de tedavinin kalitesine bağlıdır. Enzim replasman tedavisinin başlatıldığı zaman genellikle kritiktir.
Enzim replasman tedavisi, kondroitin sülfat ve dermatan sülfat gibi maddeleri parçalayabilir. Böylece hastalığın seyrini yavaşlatır. Ancak sorun, organizmada daha önce meydana gelen hasarın genellikle geri döndürülemez olmasıdır. Bu yaşam kalitesini düşürür, ancak aynı zamanda etkilenen kişinin daha erken ölmesine neden olabilir. Maroteaux-Lamy sendromunun seyri, erken ortaya çıkarsa hızlı olabilir. Hasta, uygulanan ilaca da iyi yanıt verebilir. Bu durumda, Maroteaux-Lamy sendromu buna göre yavaş ilerleyecektir.
önleme
Şimdiye kadar, Maroteaux-Lamy sendromunun gelişimi için hiçbir dış faktör bilinmemektedir. Şu anda tek önleyici tedbir, aile planlamasında genetik danışmadır.
tamamlayıcı tedavi
Maroteaux-Lamy sendromunun tedavisi karmaşık ve uzun süreli olduğu için klasik takip bakımı gerekli değildir. Aksine, etkilenenler hastalıkla güvenli bir şekilde başa çıkmaya ve zorluklara rağmen olumlu bir tutum geliştirmeye odaklanmalıdır. Gevşeme egzersizleri ve meditasyon da zihni sakinleştirmeye ve odaklanmaya yardımcı olabilir. Boy kısalığı estetikte azalma ile el ele gittiği için, herhangi bir aşağılık kompleksi ve zayıf benlik saygısı, gerekirse bir terapistle tartışılmalıdır. Bu, hastalığı daha iyi kabul etmeye ve uzun vadede yaşam kalitesini iyileştirmeye yardımcı olabilir.
Bunu kendin yapabilirsin
Maroteaux-Lamy sendromundan etkilenenler için kendi kendine yardım seçenekleri mevcut değildir. Hastalık sadece semptomatik olarak tedavi edilebilir; nedensel bir tedavi yoktur.
Hastalığın semptomlarını hafifletmek için, etkilenenler enzimlerin ve çeşitli ilaçların alımına bağlıdır. Burada düzenli ve reçeteli alım sağlanmalıdır. Ancak ağır vakalarda kalbe cerrahi müdahaleler gereklidir. Kalbi gereksiz yere zorlamamak için gereksiz efordan kaçınılmalıdır. Bu özellikle ani veya ani yükler için geçerlidir.
Hasta veya ebeveyn tekrar çocuk sahibi olmak isterse, Maroteaux-Lamy sendromunun tekrar ortaya çıkmasını önlemek için genetik danışmanlık faydalı olabilir. Yakın insanlarla veya arkadaşlarla yapılan görüşmeler genellikle psikolojik şikayetleri veya depresyonu hafifletebilir. Maroteaux-Lamy sendromlu diğer hastalarla temas genellikle hastalık üzerinde çok olumlu bir etkiye sahiptir ve etkilenen kişinin yaşam kalitesini muhtemelen iyileştirebilecek bir bilgi alışverişine katkıda bulunabilir. Bununla birlikte, sendromun tam olarak iyileştirilmesi sağlanamaz.
.jpg)
.jpg)

.jpg)
.jpg)










.jpg)


.jpg)