Meckel-Gruber sendromu (MKS) kalıtsal bir hastalıktır. Ağır doğuştan sakatlıklar ile karakterizedir. Etkilenen yenidoğanlar genellikle doğumdan sonraki ilk iki hafta içinde ölür.
Meckel-Gruber Sendromu nedir?

© hywards - stock.adobe.com
Meckel-Gruber sendromu böbrek kistleri, gelişimsel bozukluklar ve merkezi sinir sistemi bozuklukları ile karakterize kalıtsal bir hastalıktır. Hastalık da adı altında Meckel sendromu bilinen.
Almanya'da 100.000 doğumda 0.7 ila 7.5 yenidoğan hastalıktan istatistiksel olarak etkileniyor. Hastalık Finlandiya'da çok daha yaygındır. Burada 9000 yenidoğandan biri etkileniyor. Hamileliğin sona ermesi yoksa çocuklar genellikle perinatal dönemde yani yaşamın yedinci gününden önce ölürler.
nedenleri
Hastalık, otozomal resesif bir genetik bozukluk yoluyla kalıtılır. Otozomal resesif bir kalıtım modunda, genetik kusur 22 sözde otozom çiftinden biridir. Otozomlar, gonozomların aksine cinsiyet üzerinde hiçbir etkisi olmayan kromozomlardır. Meckel-Gruber sendromu cinsiyete bakılmaksızın geçmektedir. Resesif, hastalığın ancak biri babadan diğeri anneden olmak üzere iki hastalıklı gen çocuğa geçtiğinde ortaya çıkması anlamına gelir.
Bir çocuğun Meckel-Gruber sendromunu geliştirebilmesi için, çocuğun hem babasının hem de annesinin hastalığın taşıyıcısı olması gerekir. Ebeveynler, her biri yalnızca bir hastalıklı gen taşıdıklarından hiçbir belirti göstermezler. Hastalığın başlangıcı için ikinci hastalıklı gen eksiktir. Ebeveynlere ayrıca iletkenler, yani kusurlu genin taşıyıcıları da denir. Her iki ebeveyn de kondüktör ise, Meckel-Gruber sendromu gelişme olasılığı istatistiksel olarak yüzde 25'tir.
Ebeveynler akraba ise, olasılık artar. Hastalığa neden olan gen şimdiye kadar sadece kısmen bulundu. Üç farklı gen lokasyonundaki değişikliklerin hastalıktan sorumlu olduğu görülmektedir. Kromozom 17, 11 ve 8 üzerindedirler.
Belirtiler, rahatsızlıklar ve işaretler
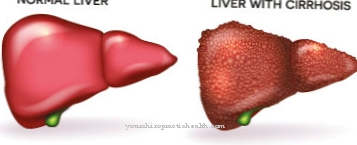
Kistik böbrekler, Meckel-Gruber sendromunun karakteristiğidir. Böbreklerde çok sayıda sıvı dolu vezikül oluşur, böylelikle böbreklerin filtre işlevi ciddi şekilde kısıtlanır. Böbrek kistlerinin oluşumu zorunludur, yani böbrek kisti yoksa Meckel-Gruber sendromu sorunu olamaz. Karaciğer kistleri de oluşabilir. Bunlar bazen karaciğer fibrozuna yol açar. Çocuklar ayrıca ensefaloselden muzdariptir.
Beyin yanlış tasarlanmış ve kafatası çoğu zaman düzgün bir şekilde kapatılmamış, böylece beynin bazı kısımları kafatasından dışarı çıkacak. Diğer beyin malformasyonları gözlemlendi. Meckel-Gruber sendromunda ağız bölgesinde bir malformasyon olan yarık dudak ve damak da oluşabilir. Çoğunlukla yenidoğanlar da mikroftalmi hastasıdır. Mikroftalmi ile gözler alışılmadık derecede küçüktür veya muhtemelen sadece ilkeldir.
Meckel-Gruber sendromunun bir başka belirtisi de polidaktili denen çoklu parmaktır. Yani ondan fazla parmak veya ondan fazla ayak parmağı var. Her iki tarafta çift başparmak özellikle yaygındır, böylece hasta on parmak yerine on iki parmağa sahiptir.
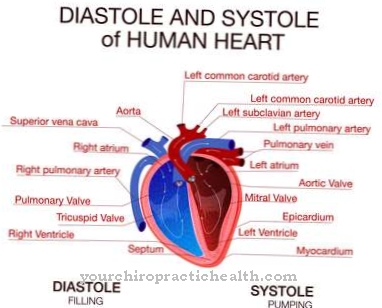
Situs inversus aynı zamanda kalıtsal hastalığın bir olgusudur. Tüm organlar vücudun diğer tarafında ters çevrilmiştir. Örneğin kalp solda ve karaciğer sağdadır. Meckel-Gruber sendromunun diğer semptomları, safra kanallarının ve az gelişmiş akciğerlerin malformasyonlarıdır.
Hastalığın teşhisi ve seyri
Kist böbreği, Meckel-Gruber sendromunun teşhisinde önemli bir ipucudur. Şap hastalığı için minimal tanı kriterleri böbrekteki kistik değişiklikler, karaciğerdeki fibrotik değişiklikler ve ensefalosel veya merkezi sinir sisteminin diğer malformasyonlarıdır. Hastalığın doğum öncesi teşhisi ultrason kullanılarak yapılır.
Hastalıklı bir fetüsün kafatasının kistik olarak değiştirilmiş bir iç kısmı ve bazen başka kafatası kusurları da vardır. Böbrekler de büyümüştür. Meckel-Gruber sendromunun bu belirtileri, gebeliğin ilk üç aylık döneminin sonunda zaten bulunabilir. Hamilelik devam ederken, diğer anormallikler sonografi ile tespit edilebilir.
Bir amniyotik sıvı testi, artmış bir alfa-fetoprotein seviyesini ortaya çıkarır. Bu kafatası ve CNS anormalliklerinden kaynaklanır ve ciddi kafatası deformitesinin kesin bir işaretidir.
Komplikasyonlar
Ne yazık ki, Meckel-Gruber sendromu nedeniyle çoğu durumda hasta doğumdan birkaç hafta sonra ölür. Bu nedenle özellikle çocuğun yakınları ve ebeveynleri ağır fiziksel şikayetlerden veya depresyondan etkilenmekte ve bu nedenle psikolojik tedaviye de ihtiyaç duymaktadır. Meckel-Gruber sendromu nedeniyle kendileri de ciddi engellerden muzdariptir ve bu nedenle hayatta kalamazlar.
Her şeyden önce, bu, hastanın böbrek ve karaciğerinde malformasyonlara, yetersizliğe ve dolayısıyla ölüme yol açar. Hastalar ayrıca yarık damaktan ve dolayısıyla yiyecek alımında kısıtlamalardan muzdariptir. Hastaların yaşam beklentisi, Meckel-Gruber sendromu tarafından dürüstçe sınırlandırılır ve azaltılır.
Meckel-Gruber sendromunu tedavi etmek veya semptomları ortadan kaldırmak maalesef mümkün değildir. Çocuklar doğumdan sonra çok erken ölüyor. Ne yazık ki, yaşamı desteklemek için başka önlemler almak mümkün değildir, bu nedenle başka komplikasyonlar yoktur. Kural olarak, ebeveynlerin çocuk öldükten sonra psikolojik tedaviye ihtiyacı vardır.
Ne zaman doktora gitmelisiniz?
Meckel-Gruber sendromundan muzdarip insanlar zaten doğumda ciddi sağlık bozuklukları gösteriyor. Bozukluklar ve arızalar genellikle doğum sürecinde tespit edilebilir. Etkilenen hastaların çoğu yarık dudak ve damakla doğar ve mümkün olan en kısa sürede tıbbi yardım almalıdır. Yatarak doğum durumunda, hemşireler ve doktorlar yenidoğanın ilk bakımını üstlenir. Çoğu zaman, çocuğun hayatta kalmasını sağlamak için acil ameliyat emri verilir. Evde doğum veya doğum merkezinde doğum durumunda ebeler ve kadın doğum uzmanları bu görevleri üstlenirler. Çocuğun en yakın hastaneye nakledilmesini organize etmek sizin sorumluluğunuzdadır.
Bu nedenle ebeveynlerin bu doğum şekillerinde aktif olmaları gerekmez. Tıbbi olarak eğitilmiş bakım personeli bulunmadan kendiliğinden doğum olması durumunda, derhal bir ambulans servisi uyarılmalıdır. Baş eğrilmiş veya deforme olmuşsa, kafatası açıksa veya uzuvlar düzensizse acilen doktora ihtiyaç vardır. Sendrom, ondan fazla parmak veya ayak parmağının varlığı ile karakterizedir. Solunum bozuksa, bebeğin hayatının ilk dakikalarında ölmemesi için acil doktor gelene kadar ek ilk yardım önlemleri alınması gerekir. Sağkalım ancak ağızdan ağza resüsitasyon ile sağlanabilir.
Terapi ve Tedavi
Meckel-Gruber sendromu tedavi edilemez. Doğumdan önce teşhis konulursa, genellikle gebeliğin sonlandırılması düşünülür. Kafatasının ve organların ciddi malformasyonları nedeniyle, Meckel-Gruber sendromundaki ölüm oranı yüzde 100'dür, bu da etkilenen tüm yenidoğanların uzun vadede yaşayamayacağı anlamına gelir. Çoğu çocuk ilk yedi gün içinde ölür; hiçbir çocuk genellikle iki haftadan fazla yaşamaz.
Görünüm ve tahmin
Meckel-Gruber sendromunun prognozu son derece olumsuzdur. Sendrom, genetik bir kusura dayanmaktadır. Bu, mevcut tıbbi olanaklarla tedavi edilemez. Çocuk ciddi engellerle doğar ve yaşama şansı çok azdır. Yasal gereklilikler nedeniyle, doktorların ve araştırmacıların insan genetiğini hiçbir şekilde değiştirmelerine izin verilmiyor. Sonuç olarak, doktorlar yalnızca çeşitli semptomlardan kurtulmayı sağlayan tedavi seçeneklerini kullanmaya odaklanabilir.
Bununla birlikte, bu hastalık için çok sayıda ciddi sağlık kısıtlamasının belgelenmesi gerektiğinden, mevcut tedavi seçenekleri, etkilenen kişiyi stabilize etmek için yeterli değildir. Doğumdan sonraki birkaç gün veya hafta içinde, önceden bilinen tüm Meckel-Gruber sendromu vakaları, hastanın erken ölümüne yol açar. Teşhisin ne kadar erken konulduğuna ve ne kadar hızlı tıbbi önlemlerin alındığına bakılmaksızın, etkilenen kişinin ortalama yaşam beklentisi doğumdan sonra bir ila iki hafta arasındadır.
Fiziksel malformasyonlar iskelet sistemi ve organların çeşitli alanlarını etkiler. Yenidoğanın vücudu, organizmayı stabilize etmek için gerekli olan sayısız operasyondan kurtulamayacak kadar zayıftır. Dolayısıyla tüm çabalara rağmen organ yetmezliği ve dolayısıyla erken ölüm kaçınılmaz olarak ortaya çıkacaktır.
önleme
Prensip olarak, Meckel-Gruber sendromu önlenemez. Erken teşhis hastalığı engellemez, sadece gebeliğin daha erken sonlandırılmasını sağlar. Araştırmacılar, ciddi kalıtsal hastalıktan sorumlu değişikliklerin olabileceği üç genetik konum belirlediler. Bu konumlar, FMD lokasyonları olarak bilinir:
- MKS1, kromozom 17 üzerindedir
- MKS2, kromozom 11 üzerinde
- Sekizinci kromozomda MKS3.
Pakistan'daki insanlarda FMD3 genindeki değişiklikler tespit edildi. FMD1 genindeki değişiklikler Finlandiya ve Avrupa'da da meydana geldi. Şimdiye kadar FMD1 genindeki bir değişiklik, hastalığın kesin bir nedeni olarak tanımlanmıştır. Bu kusurlu genin varlığını kontrol eden genetik bir teşhis var. Bu teşhisin gerçekleştirilmesi için ön koşul, Meckel-Gruber sendromunun güvenilir bir teşhisinin zaten yapılmış olmasıdır.
Meckel-Gruber sendromu bu hasta çocukta genetik teşhis ile de tespit edilebiliyorsa, daha sonraki gebeliklerde doğum öncesi genetik teşhis yapılabilir. Bu şekilde ebeveynlere, doğmamış çocuklarının genetik kusurun taşıyıcısı olup olmadığı kesin olarak söylenebilir. FMD'den şüpheleniyorlarsa ebeveynleri test etmek de mümkündür. Genetik kusurun taşıyıcısı olup olmadığınızı ve hastalığı gelecekteki yavrulara geçirme riskinin olup olmadığını belirlemek için bir kan testi yapılır.
tamamlayıcı tedavi
Kural olarak, Meckel-Gruber sendromu aynı zamanda çocukların yaşam beklentisini de önemli ölçüde azaltır, böylece doğumdan sadece birkaç hafta sonra ölürler. Takip bakımı, yaslı olana odaklanır.
Bazen profesyonel psikolojik destek, çocuğun kaybı ve kederi ile başa çıkmaya yardımcı olabilir. Etkilenenlerle birlikte bu, şiddetli psikolojik stresi absorbe etmek için terapötik önlemler geliştirir. Halihazırda etkilenmiş olan ebeveynler tekrar çocuk sahibi olmak isterlerse, önceden başka bir hasta çocuğun olasılığını belirlemek için tedavi eden jinekoloğa danışmalarını tavsiye ederiz.
Bunu kendin yapabilirsin
Meckel-Gruber sendromu, vakaların büyük çoğunluğunda çocuğun ölümüne yol açan ciddi bir hastalıktır. Bu olumsuz prognoz nedeniyle, en önemli kendi kendine yardım ölçüsü, terapötik destek aramaktır. Jinekolog, ebeveynleri, tipik korku ve endişelerin tartışılabileceği ve çözülebileceği uygun bir uzmana yönlendirebilir. Bir kendi kendine yardım grubuna katılmak da tavsiye edilir. Etkilenen diğer akrabalarla konuşmak, hastalıkla ve çoğunlukla olumsuz sonuçlarıyla baş etmeyi kolaylaştırır.
Sorumlu jinekologla birlikte, çocuğun terime mi yoksa kürtajla mı taşınacağına karar verilmelidir. Genellikle ebeveynler kürtaj yapmaya karar verir çünkü iyileşme şansı çok düşüktür, ancak bazı durumlarda normal doğum da mümkündür ve mantıklıdır. Alternatif olarak palyatif doğum vardır.
Ebeveynler ne seçerse seçsinler, psikolojik desteğe ve arkadaş ve akrabaların yardımına ihtiyaç vardır. Kadın daha sonra tekrar gebe kalmak isterse, sağlıklı bir çocuk sahibi olma şansını belirlemek için kapsamlı bir muayene yapılmalıdır.

.jpg)











.jpg)


.jpg)