Holt-Oram Sendromu öncelikle kalp kusurları ve başparmak anomalileri ile ilişkili olan ve bir mutasyondan kaynaklanan bir malformasyon sendromudur. Çoğu durumda, nedensel mutasyon ara sıra meydana gelir ve bu nedenle yeni bir mutasyona karşılık gelir. Kalp kusurunun cerrahi olarak düzeltilmesi tedavinin odak noktasıdır.
Holt-Oram Sendromu?

© SmirkDingo - stock.adobe.com
Ekstremitelerin ağırlıklı tutulduğu konjenital malformasyon sendromları, kol ve bacaklarda çeşitli deformiteleri içeren bir grup hastalıktır. Bunlardan biri bu Holt-Oram Sendromu, olarak da adlandırılır Kalp-el sendromu bilinen. Sendrom, üst ekstremitelerde ve kalpte malformasyonlar ile konjenital hastalıkları içeren atriyodijital displaziler grubuyla ilişkilidir.
Holt-Oram sendromuna ek olarak, kalp-el sendromu tip 2, kalp-el sendromu tip 3 ve Sloven tipi kalp-el sendromu bu hastalık grubuna aittir. Holt-Oram sendromu ilk olarak 1960 yılında tanımlanmıştır. İngiliz pediatrist Holt ve kardiyolog Samuel Oram, hastalığı tanımlayan ilk kişilerdir. Holt-Oram sendromu Bu nispeten nadir bir hastalıktır ve 100.000 kişide etkilenen bir kişinin ortalama yaygınlığı ile ilişkilidir.
Holt-Oram sendromunda ellerdeki ve kalpteki malformasyonların nedeni genetiktir. Sendromun semptomları, şüpheli nedenleri kadar çeşitlidir. Hastalıktaki malformasyonlar kalp ve başparmağa yoğunlaşır.
nedenleri
Holt-Oram sendromu genetik ve doğuştan bir hastalık olmasına rağmen, şimdiye kadar belgelenen vakalarda hemen hemen hiç ailevi sıklık yoktur. Sendrom, bireysel vakalarda otozomal dominant bir kalıtım tarzında kalıtsal görünse de, vaka belgelerinin büyük bir kısmı onun sporadik oluşumunu göstermektedir. Belgelenen vakaların yaklaşık yüzde 85'i yeni bir mutasyondan kaynaklanıyor gibi görünüyor.
Holt-Oram sendromunun birincil nedeni, 12q23-24 gen lokusundaki genetik mutasyonlarda yatmaktadır. Kodlanmış kalp gelişimi. Proteinin kesin işlevleri henüz açıklığa kavuşmadı. Hamilelik sırasında annede toksinlere maruz kalma veya yetersiz beslenme gibi dış faktörlerin TBX5 geninin mutasyonunu destekleyip desteklemediği henüz belirlenmemiştir.
Gendeki mutasyonlar, Holt-Oram sendromlu 100 hastadan en az 70 kişide tespit edilebilir. Bununla birlikte bilim, diğer genlerdeki anormalliklerin de sendromun semptomlarına neden olabileceğini varsayar. Örneğin, malformasyon sendromu, üç taraflı başparmak polisindaktili ile ilişkilidir.
Belirtiler, rahatsızlıklar ve işaretler
Holt-Osram sendromlu hastalar, öncelikle başparmak ve kalbi etkileyen bir dizi malformasyondan muzdariptir. Hastanın malformasyonlarının lokalizasyonu yaygın olmakla birlikte, kalpte ve baş parmakta farklı tipte malformasyonlar mümkündür. Klinik tablo bu nedenle oldukça çeşitlidir.
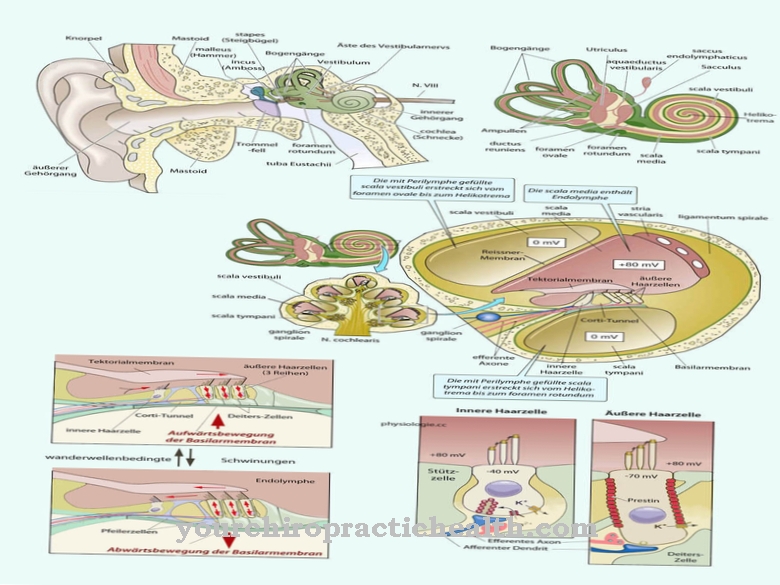
Kalp kusurları, örneğin bir ventriküler septal kusur, bir atriyal septal kusur, bir kardiyak aritmi veya bir iletim bozukluğu şeklinde kendini gösterebilir. İskelet anomalileri, başparmakların azalma bozukluklarına karşılık gelebilir, ancak parmaklığın uygulanmaması gibi anormallikler de ortaya çıkar.
Sendromlu birçok hasta ayrıca radioulnar sinostozlardan ve kaburga, kürek kemiği veya klavikula anormalliklerinden muzdariptir. Ayrıca Holt-Oram sendromu, pektus karinatum ve skolyoz ile ilişkilidir. Etkilenenlerin çoğunluğu ayrıca el veya ayak parmaklarında sindaktiliden muzdariptir. Bireysel durumlarda bu semptomlar hipertelorizm ile ilişkilidir.
Hastalığın teşhisi ve seyri
Holt-Oram sendromu sıklıkla yanlış teşhis edilir. Ayırıcı tanıda, doktor aynı kol malformasyonları ve kalp kusurları ile ilişkili olan kromozom 20 üzerindeki SALL4 genindeki gen mutasyonlarından sonra semptom kompleksini Okihiro sendromundan ayırmalıdır. Ayırıcı tanıda özellikle önemli olan, Okihiro sendromlu hastaların genellikle bir Duane anomalisine sahip olmasıdır.
Gözlerini kısarlar, genellikle böbrek malformasyonlarından etkilenirler ve işitme bozuklukları, ayak anomalileri veya kulak bozuklukları vardır. Trombositopeni-Radius-Yok-Sendromu, esas olarak laboratuvar tanıları yoluyla elde edilen Holt-Oram sendromundan da ayırt edilmelidir. Klinik olarak benzer bir tabloya sahip diğer klinik tablolar Fanconi anemisi ve Pallister Hall sendromudur.
Holt-Oram sendromlu hastaların yaşam beklentisi ortalamanın altında değildir. Sadece ağır vakalarda, prognozu elverişsiz kılan, tedavi edilmesi zor bir kalp kusuru vardır.
Komplikasyonlar
Holt-Oram sendromu, hastada yaşamı ve günlük yaşamı zorlaştırabilecek bir dizi farklı malformasyon ve deformiteye neden olur. Her şeyden önce, kalp malformasyonlardan etkilenir, böylece hasta bir kalp kusurundan muzdariptir. Ayrıca, etkilenen kişinin en kötü durumda ölebileceği bir kardiyak aritmi de vardır.
Başparmaklarda da anormallikler meydana gelir, bu nedenle günlük yaşamdaki belirli hareketler veya süreçler de zorlaşır. Vücuttaki deformitelerin diğer çocukların alay ve zorbalığına yol açması, birçok hastada psikolojik şikayetlere ve depresyona yol açması nadir değildir. En kötü durumda böbrek yetmezliğine yol açabilen böbrek rahatsızlıklarının ortaya çıkması nadir değildir.
Ayrıca, etkilenenler ayrıca bir görme ve işitme bozukluğundan muzdariptir. Kural olarak, ölüme yol açan kalp kusuru olmadığı sürece, Holt-Oram sendromunun bir sonucu olarak yaşam beklentisi değişmeden kalır. Holt-Oram sendromunun nedensel tedavisi genellikle mümkün değildir, bu nedenle yalnızca semptomlar tedavi edilebilir. Çoğu durumda psikolojik destek de gereklidir.
Ne zaman doktora gitmelisiniz?
Holt-Oram sendromu genellikle doğumdan kısa bir süre sonra teşhis edilir. Malformasyonların ne kadar şiddetli olduğuna bağlı olarak, etkilenen çocuğun daha fazla tıbbi muayeneye ihtiyacı olabilir. Prensip olarak, ciddi ikincil hastalık riskini azaltmak için kalp kusurları derhal tedavi edilmelidir. Kardiyak aritmi gibi komplikasyonlar veya atriyal septal defekt belirtileri gelişirse, tıbbi tavsiye gereklidir. İskelet başparmağı anormallikleri için bir tıp uzmanına da danışılmalıdır.
Etkilenen çocukların ebeveynleri, doktorlarına yakından danışmalı ve onları olağandışı semptomlar konusunda bilgilendirmelidir. Holt-Oram sendromu kalıtsal bir hastalık olduğu için nedensel tedavi mümkün değildir. Bu nedenle, hangi malformasyonların meydana geldiğine ve hastanın bünyesine bağlı olarak, hastaların ömür boyu tedavi edilmesi gerekebilir. Bu genellikle duygusal şikayetlere de neden olduğundan, eşlik eden psikolojik destek endikedir. Zorbalık veya alaydan muzdarip çocuklar, ebeveynleriyle terapötik danışmanlık almalıdır.
Bölgenizdeki doktorlar ve terapistler
Terapi ve Tedavi
Holt-Oram sendromlu hastalar için nedensel tedavi mevcut değildir. Gelecekte nedensel bir tedavi için umut var, çünkü gen terapisi şu anda tıbbi araştırmanın konusu. Bununla birlikte, bu tür bir terapi klinik aşamaya ulaşmadığı sürece, Holt-Oram sendromu tedavi edilemez bir hastalık olarak kalır.
Şu anda hastaları tedavi etmek için yalnızca semptomatik tedavi seçenekleri mevcuttur. Terapi, bireysel vakadaki semptomlara dayanmaktadır. Kalp kusurunun erken düzeltilmesi özellikle önemlidir. Bu düzeltme ameliyatla yapılır. Vohof septal defekti durumunda, cerrahi prosedür söz konusu defekti kapatmayı amaçlamaktadır. Aynısı ventriküler septal defekt için de geçerlidir.
Ekstremitelerdeki malformasyonların düzeltilmesi başlangıçta ikincil öneme sahiptir. Kalp kusurunu başarıyla düzelttikten sonra, rekonstrüktif cerrahi prosedürler eksik parmak tellerini ve ayrı sindaktilileri geri yükleyebilir. Mevcut bir skolyozla genellikle fizyoterapötik bakım altında mücadele edilir. Özellikle şiddetli vakalarda, bir titanyum kuşak protezi implante etmek için ameliyat gerekli olabilir.
Çoğu durumda, pectus carinatum için herhangi bir müdahale gerekmez. Bununla birlikte, psikolojik nedenlerden ötürü, göğüs kafesi, örneğin Nuss prosedürünün ardından cerrahi olarak yeniden şekillendirilebilir.
Görünüm ve tahmin
Holt-Oram sendromunun prognozu olumludur. Genetik bir kusur olmasına rağmen mevcut tıbbi seçeneklerle yeterince tedavi edilebilir. Sendromlu bir kişinin yaşam beklentisi çoğu durumda ortalamanın altında değildir. Ciddi bir malformasyon durumunda, yaşam beklentisinde önemli kayıplar olabilir. Bu hastalarda prognoz açıkça kötüleşmiştir. Kalbin aktivitesi sınırlıdır ve yaşamın erken sona ermesine neden olabilir.
Ancak hastaların çoğu iyi ve başarılı bir şekilde tedavi edilebilir. Mevcut genetik kusur nedeniyle tedavi olmasa da, çeşitli düzeltme seçenekleri için iyi beklentiler vardır. Kalbin aktivitesi düzenlenir ve mümkünse cerrahi bir prosedürde tamamen düzeltilir. Sağlıklı insanlara göre yaşam tarzında kalıcı bir bozulma olsa da tedavi sayesinde iyi bir yaşam kalitesi elde edilir.
Fiziksel anormallikler veya malformasyonlar bir sonraki adımda değiştirilir. Genellikle çocuğun büyüme aşaması tamamlandıktan sonra, mevcut malformasyonların gerekli veya istenen bir şekilde düzeltilmesine başlanır. Gelişim sürecindeki aksaklıklar önemli bozukluklara yol açıyorsa, semptomları hafifletmek için çocukluk döneminde düzeltici önlemler alınır. Optik değişiklikler nedeniyle hasta psikolojik sonuçlar yaşayabilir. Bu genel prognozu kötüleştirir.
önleme
Şu ana kadar Holt-Oram sendromu önlenemiyor çünkü dışsal etki faktörleri kesin olarak açıklığa kavuşmadı.
tamamlayıcı tedavi
Holt-Oram sendromu doğuştan bir hastalık olduğu için tam olarak tedavi edilemez. Bu nedenle, takip bakımının önlemleri veya olanakları çok sınırlıdır, bu nedenle etkilenen kişi öncelikle erken teşhis ve sonraki tedaviye bağımlıdır. Hasta veya ebeveyn çocuk sahibi olmak istiyorsa bu sendromun tekrar oluşmaması için genetik danışmanlık verilir.
Holt-Oram sendromunun tedavisi öncelikle kalp kusurunun tedavisine yöneliktir. Bu, hastanın işlemden sonra iyileşmesi ve dinlenmesi gereken cerrahi bir prosedürle düzeltilir. Efor veya fiziksel aktiviteden kaçınılmalıdır. Fizyoterapötik tedavinin gerekli olması alışılmadık bir durum değildir ve egzersizlerin çoğu kendi evinizde de yapılabilir.
Etkilenenler bazen kendi ailelerinin ve arkadaşlarının yardımına ve desteğine bağımlıdır. Bu aynı zamanda psikolojik rahatsızlıkları veya depresyonu da önleyebilir. Dahası, sağlıklı bir diyetle sağlıklı bir yaşam tarzı, Holt-Oram sendromunun seyri üzerinde çok olumlu bir etkiye sahiptir.
Bunu kendin yapabilirsin
Holt-Oram sendromu önlenemez ve kendi kendine yardım yoluyla da tedavi edilemez. Bu sendromla, etkilenenler, hastanın yaşam beklentisini uzatmak için her zaman kalp kusurunu tedavi etmek için bir operasyona güvenirler. Sendrom ne kadar erken fark edilirse, hastalığın olumlu seyri olasılığı o kadar yüksek olur. Vücuttaki diğer şekil bozuklukları da cerrahi olarak düzeltilmelidir.
Etkilenenlerin çoğu aynı zamanda psikolojik şikayetlerden veya bu sendromla ilişkili aşağılık komplekslerinden muzdarip olduğu için, psikolojik tedaviye bağımlıdırlar. Ancak diğer hastalarla, kendi ebeveynlerinizle veya arkadaşlarınızla yapılan görüşmeler de hastanın özgüvenini güçlendirebilir ve böylece psikolojik şikayetleri hafifletebilir. Dahası, bazı hastalar günlük yaşamlarında hemcinslerinin yardımına bağımlıdır, bu nedenle sıcak bakım, Holt-Oram sendromunun seyri üzerinde çok olumlu bir etkiye sahiptir.
Hastalık iç organları da etkileyebileceğinden, hastalar çeşitli doktorlar tarafından düzenli muayene ve kontrollere bağımlıdır. Bu, örneğin böbrek problemlerini önleyebilir. Etkilenen çocuklar, hastalığın sonuçları ve komplikasyonları hakkında bilgilendirilmelidir.
.jpg)

.jpg)



.jpg)